This was the second review article I wrote for my Fall 2008 Frontiers in Chemical Biology class at MIT. Download a PDF copy here.
Summary of Recent Advances
Despite hundreds of years of development, the basic strategy of synthetic organic chemistry - convergent generation of a target molecule from simpler starting materials - has remained largely unchanged. In the modern age of systems biology and high-throughput genomics / proteomics, the pace of such a strategy is insufficient to meet the growing demand for biologically active compounds. Building upon the goals of combinatorial chemistry (a largely failed attempt to address this issue), the emerging method of diversity-oriented synthesis (DOS) is poised to revolutionize the discovery and development of new pharmaceuticals. Arising from the intersection of chemistry and biology, DOS combines the structural diversity of natural products with the transformative power of synthetic chemistry to rapidly interrogate larger expanses of biologically-active chemical space than ever before possible.
Introduction
Perhaps the most significant contribution from the field of synthetic organic chemistry is the improvement of human healthy through the generation of biologically active compounds and pharmaceuticals. The development of a host of powerful reactions, reagents, and techniques has permitted the efficient synthesis of an ever-increasing number of such molecules. As our capabilities have grown, so has our ambition, particularly with regard to the synthesis of complex natural products. The therapeutic value of such products has been known since ancient times (in the guise of medicinal herbs), but only through the tools of modern chemistry has the identification and artificial production of the active compounds been a possibility. The synthesis of such compounds is complicated by a variety of structural features, most notably the large number of heteroatoms (which often necessitate protecting groups) and stereocenters (which often require difficult stereoselective reactions or separations). Therefore, the discovery of new techniques for the synthesis of such compounds is one of the most active areas of research at the frontier of chemical biology.
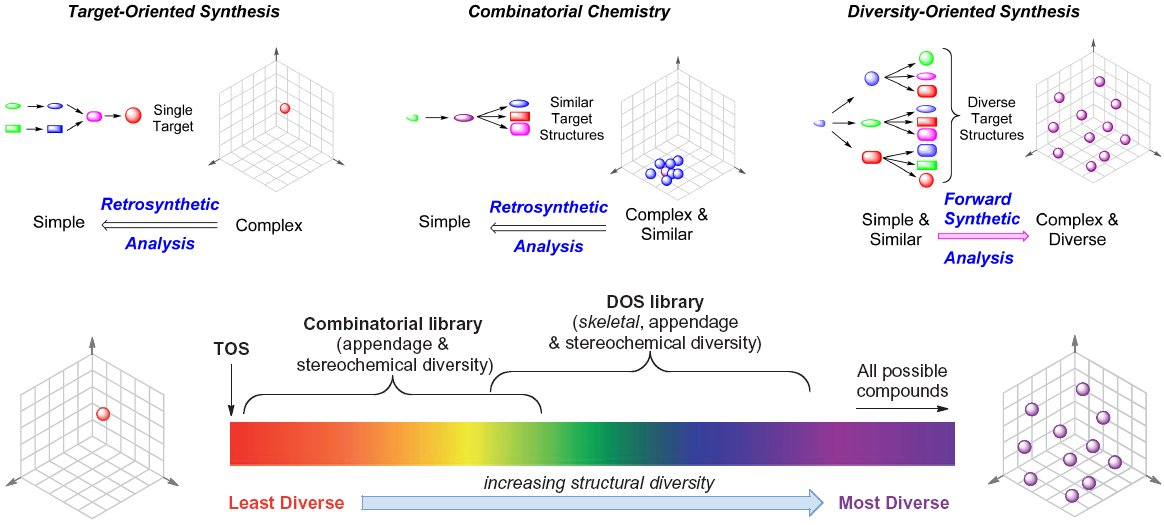
The strategies typically employed in synthetic chemistry can be broadly classified into three approaches that are distinguished by chronology, philosophy, and coverage of chemical space (an abstract concept that treats the set of all possible synthetic molecules as a three-dimensional space). These can most easily be represented and understood visually, as demonstrated in Figure 1 - Comparison of Target-Oriented, Combinatorial, and Diversity-Oriented Syntheses [This figure illustrates the philosophy behind each of the three categories of synthetic methodologies. Target-oriented synthesis and combinatorial chemistry use retrosynthetic analysis to interrogate a single point in chemical space or its immediate surroundings, respecitvely, whereas diversity-oriented synthesis tries to maximize skeletal diversity and thus coverage of chemical space. This figure is adapted from [3*]].
{kind=link}
The simplest approach is known as target-oriented synthesis (TOS), and is just classic synthetic chemistry in which a single compound of interest is identified and synthesized. It is the oldest and most well established method, and has the benefit of a powerful planning algorithm known as retrosynthetic analysis [1]. It is also the least diverse and least scalable method, by definition covering only a single point (molecule) in chemical space for each synthetic scheme employed. TOS is still an essential part of the chemist's repertoire when the compound of interest is known, but its time- and labor-intensive nature makes it poorly suited for the discovery of new compounds.
Within the past few decades, chemists have attempted to overcome these limitations through the development of an approach known as combinatorial chemistry. The hallmark of combinatorial chemistry is the synthesis of large libraries - sometimes with tens of thousands of members - of compounds that can then be screened for biological activity. Combinatorial chemistry was facilitated by the development of advanced microscale and high-throughput synthetic/analytical techniques, and promised to revolutionize the pharmaceutical industry by greatly increasing the amount of chemical space interrogated by a single scheme. Such claims have proven largely inaccurate, as combinatorial libraries typically focus on changing only the stereochemistry or "appendage" architecture of a given moiety, and thus do not achieve true structural diversity; rather, they only expand beyond TOS to the chemical space immediately surrounding the underlying synthetic skeleton [2].
The most recent (and currently most promising) attempt to improve upon the shortcomings of combinatorial chemistry is an approach known as diversity-oriented synthesis (DOS), which aims to correct the fundamental flaw in combinatorial chemistry by producing large variations (that is, high diversity) in the skeletons of synthetic building blocks. DOS is best understood as iterative combinatorial chemistry: a library of molecules with similar skeletons is synthesized as in traditional combinatorial chemistry, and those compounds are then used as substrates for the subsequent generation of new libraries. In this way, a single DOS scheme can encompass exponentially more chemical space than either TOS or combinatorial chemistry [3*]. A variety of methods have been employed to achieve such diversity, and the most successful have come from the interface of chemistry and biology.
Biomolecules as Scaffolds
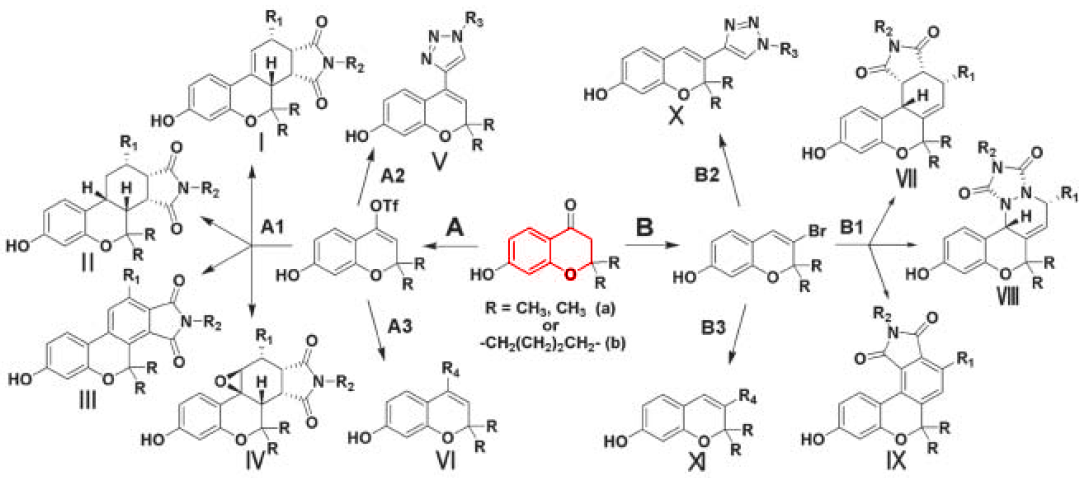
One of the fundamental hurdles in the process of drug development is the identification of skeletons that have desirable biological activity [4]. Once such so-called "hits" are found, there are a number of techniques for systematically optimizing their activity, but the actual discovery process is often based on trial and error. Chemical biology has provided a method for systematizing this process by using biological molecules (in part or in whole) as scaffolds for DOS. The logic behind this technique is that such molecules possess "privileged motifs" - structural features that have been selected and conserved by evolution - and are therefore predisposed to reside in the biologically active realm of chemical space [5]. In what will certainly become a classic in the field of DOS, Ko et al. identified the privileged motif of benzopyran, a bicyclic heterocycle that is found repeatedly in nature and is known to bind to a variety of biopolymers, as an excellent substrate for DOS. By subjecting a small number of functionalized benzopyran rings to multiple iterations of well-defined transformations (e.g. Diels-Alder, Stille, or click chemsitry), they generated a library of 22 molecules (not all illustrated) with high skeletal diversity around the benzopyran, as shown in Figure 2 - Skeletal Diversity Generated from DOS on a Privileged Benzopyran Motif [This figure illustrates the branching and iterative synthetic scheme used to generate a small library of diverse skeletons starting from a single compound bearing the benzopyran "privileged motif" (highlighted in red). Reagents and reaction conditions for each transformation are available from the appropriate reference. This figure is adapted from [6*]].
{kind=link}
Subsequent in vitro assays of these 22 compounds for revealed a range of biological activity against a human breast cancer line, with more than one-hundred-fold variation from least to most effective. This work clearly demonstrated the efficiency of DOS over traditional combinatorial chemistry by achieving appreciable biological activity with a relatively small library, and confirmed the logic of using biomolecular scaffolds. Similar strategies have recently been successfully applied to privileged motifs featuring more stereocenters and heteroatoms [7], and they will no doubt continue to be expanded upon.
Stereoselectivity through DOS
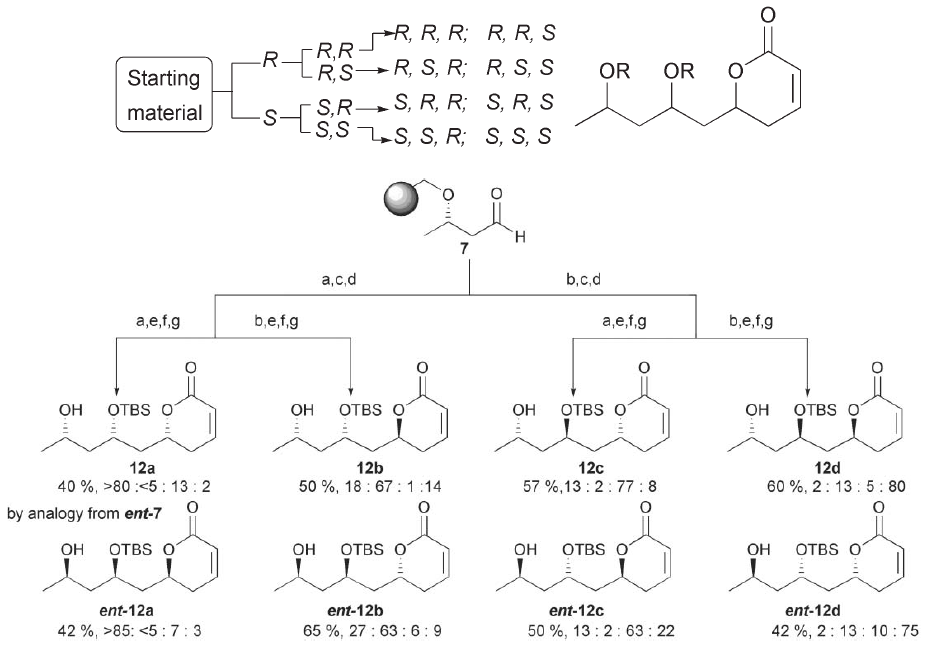
It is difficult to overstate the importance of stereochemistry in biology and synthetic chemistry. Stereochemistry is highly conserved in living systems (amino acids, saccharides, etc.), stereoisomers can have widely varying biological activity (cis-platin vs. trans-platin), and several Nobel Prizes have been awarded for stereoselective reactions (Knowles, Sharpless, etc.). Natural products are notorious for having large numbers of stereocenters, and the number of possible isomers grows exponentially with the number of chiral centers. The generation of stereoisomers is therefore important at both levels of DOS, as it can contribute to initial appendage diversity and ultimate skeleton diversity. However, the synthetic toolkit for selectively generating specific enantiomers/diastereomers is limited and can hinder the development of structure-activity relationships for all stereocenters in a molecule by making it impractical to synthesize all stereoisomers. New additions to this toolkit for use in DOS can be divided into two related categories: stereoselective reactions that generate only one stereoisomer, and stereocomplimentary reactions that generate all stereoisomers in such a way that they can be separated/isolated. Blay et al. provided a powerful addition to the first category by developing a diastereoselective Michael addition and demonstrating its ability to produce a small library of enantiomerically pure compounds starting from several achiral diketones [8]. Garcia et al. took this a step further with an addition to the second category, demonstrating a stereocomplimentary allylation that functioned on solid support (a desirable feature that greatly simplifies isolation/purification). They then validated the reaction by using it to isolate all eight isomers of the natural product cryptocarya diacetate, as shown in Figure 3 - Generation of all Isomers of Cryptocarya Diacetate via Stereocomplimentary Allylation [This figure illustrates the synthetic scheme to generate all eight isomers of the natural product skeleton shown using successive allylation reactions on a bead-bound substrate. Percent yields are shown for each product, and the reagents and reaction conditions for each transformation are available from the appropriate reference. This figure is adapted from [9**]].
{kind=link}
Other recently developed techniques include a method for stereoselective generation of glycosides (highly valuable for DOS due to their density of heteroatoms) via gold catalysis [10], and a microwave-controlled stereoselective tandem Wittig-intramollecular-Diels-Alder reaction (useful for simultaneously generating stereochemical and skeletal diversity) [11]. In both cases the authors demonstrated the ability of their method to generate both appendage and skeleton diversity, revealing the power of stereochemistry in DOS.
Biology-Oriented Synthesis
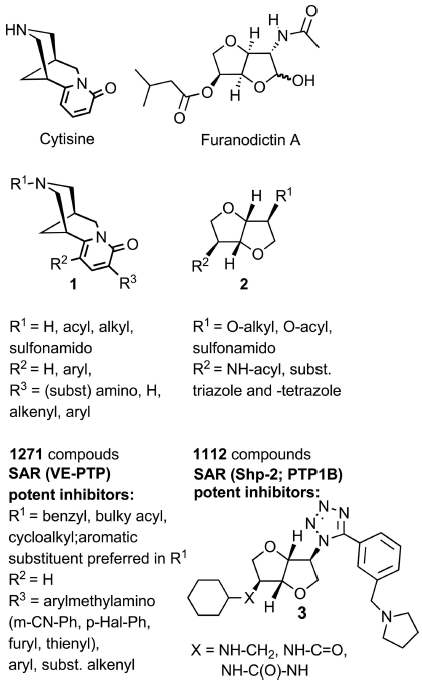
Perhaps the greatest strength of traditional synthetic methods (TOS and combinatorial chemistry) is the existence of retrosynthetic analysis for guidance. This technique allows for the design of logical and efficient syntheses prior to performing any reactions by breaking complex target molecules into simple building blocks. Despite its many strengths, DOS does not have a consensus method for the vague process of "forward synthetic analysis". One promising possibility is biology-oriented synthesis (BOS), which can be thought of as a combination of biomolecular scaffolds with stereoselective and/or biologically based transformations. The synthetic schemes and compound libraries that result from BOS are essentially guaranteed to have biological activity because they interrogate only that region of chemical space known to occur in nature. In other words, BOS trades a small amount of chemical space diversity for a greater assurance of biological function. In one of the first examples of the power of BOS, Noren-Muller et al. synthesized a large library of natural-product-inspired compounds and assayed their activity as inhibitors of clinically-relevant human phosphatases, as outlined in Figure 4 -Biology-Oriented Synthesis of Novel Phosphatase Inhibitors [This figure outlines the discovery of novel phosphatase inhibitors through BOS. The natural products at the top were used to generate the natural-product-inspired skeletons shown, and these were subjected to DOS to yield libraries of potential inhibitors. In vitro assay screens revealed the structure-activity relationships (SAR) for the four resulting classes of inhibitors, two of which are shown. This figure is adapted from [12*]].
{kind=link}
This work resulted in the discovery of four novel classes of phosphatase inhibitors, two of which were the first inhibitors for their respective phosphatases. Subsequent analysis of these compounds produced detailed structure-activity relationships that gave insight into the mechanism of the phosphatases, and validated BOS as a method for guiding drug discovery. Since this "proof of concept", a number of other groups have used BOS for various purposes. Sohma et al. developed a "click peptide" method that used an O-acyl peptide derivative to stereoselectively synthesize "difficult sequence" polypeptides with low solubility domains, and they applied this method to BOS to synthesize amyloid β analogues that could be used for Alzheimer's disease research [13]. Starting with a collection of natural-product-inspired aldehydes on solid support, Umarye et al. used stereospecific allylation to simultaneously introduce numerous stereocenters and dense functionality into the basic skeletons, again demonstrating the ability of BOS to optimize DOS [14].
Case Study: Anti-MRSA Compounds
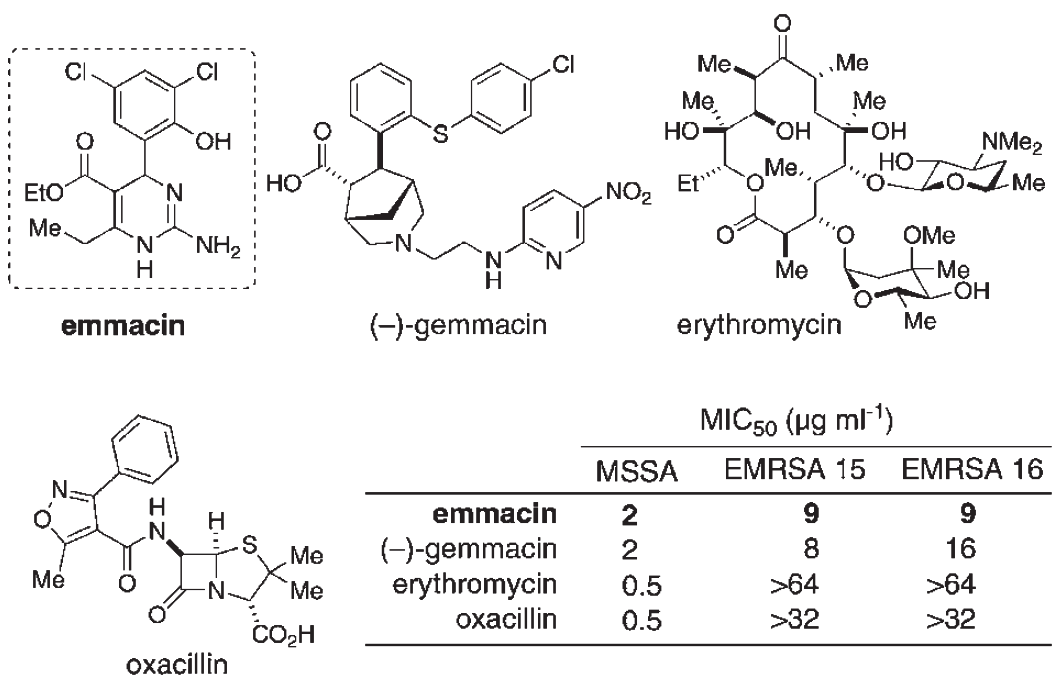
The most stringent test of a drug discovery method (and the one that traditional combinatorial chemistry has repeatedly failed) is to demonstrate that it can produce a novel potential pharmaceutical. In the case of DOS, this can be done by looking at a "case study" from the laboratory of D.R. Spring. Using DOS on a simple, easily-purified substrate, his group first generated a library of 223 small molecules to interrogate chemical space, from which they discovered antibacterial activity against the deadly methicillin-resistant Staphylococcus aureus (MRSA) [15]. They then targeted this activity by using BOS (starting with a library of natural-product-inspired compounds and using stereoselective transformations) to generate a library of 242 drug-like compounds, from which they identified a novel class of antibiotics - dihydrofolate reductase inhibitors. This class included two compounds with anti-MRSA activity significantly greater than the common antibiotics erythromycin and oxacillin, as seen in Figure 5 - Discovery of Novel Antibiotics with Anti-MRSA Activity [16] [This figure gives the structures of two members (emmacin and gemmacin) of a novel class of antibiotics (dihydrofolate reductase inhibitors) that was identified using DOS. Both compounds exhibited greater activity against MRSA than the traditional antibiotics erythromycin and oxacillin. MSSA is methicillin-sensitive Staphylococcus aureus, EMRSA-15 and -16 are two strains of MRSA, and MIC50 means the minimum dose necessary to inhibit growth of 50% of the population. This figure is adapted from [17**]].
{kind=link}
This case study is notable because it covers several of the initial steps in drug discovery, from hit detection through structure-activity relationship determination, and demonstrates the possibility for DOS to enhance all aspects of the procedure. Given the dire need for new classes of antibiotics, it is likely that this work will see continued development to the clinical phase, which would be the ultimate validation of DOS.
Conclusions
The process of drug discovery, particularly when inspired by or targeted toward natural products, is tedious and difficult. While our abilities to synthesize ever-more complex molecules has grown by leaps and bounds, our ability to investigate large swaths of chemical space in a reasonable amount of time has been relatively stagnant. The promise of combinatorial chemistry has not been realized in the decades since its inception, but a new technique offers the potential to fulfill these promises and more. By applying the tools of synthetic chemistry to the structures of biology in novel ways, DOS has the power to interrogate biologically-active chemical space with a throughput and density of coverage never before possible. Active and exciting research in this nascent field has already generated a number of surprising and promising outcomes. Continued development of reagents and techniques will make DOS one of the most popular aspects of chemical biology in the 21st century.
References
* denotes an article of special interest and ** denotes an article of outstanding interest
- Schreiber SL: Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287: 1964-1969.
- Spring DR: Diversity-oriented synthesis; a challenge for synthetic chemists. Org Biomol Chem 2003, 1: 3867-3870.
-
* Spandl RJ, Diaz-Gavilan M, O'Connell KMG, Thomas GI, Spring DR: Diversity-oriented synthesis. Chem Rec 2008, 8: 129-142.
This is the most thorough and up-to-date review available for DOS. It provides an outstanding conceptual defintion of DOS, clearly explains the rationale behind its development, and offers examples of the fields of research most likely to benefit from it.
- Tolliday N, Clemons PA, Ferraiolo P, Koehler AN, Lewis TA, Li X, Schreiber SL, Gerhard DS, Eliasof S: Small molecules, big players: the National Cancer Institute's Initiative for Chemical Genetics. Cancer Res 2006, 66(18): 8935-8932.
- Spandl RJ, Benderb A, Spring DR: Diversity-oriented synthesis; a spectrum of approaches and results. Org Biomol Chem 2008, 6: 1149-1158.
-
*Ko SK, Jang HJ, Kim E, Park SB: Concise and diversity-oriented synthesis of novel scaffolds embedded with privileged benzopyran motif. Chem Comm 2006: 2962-2964.
This is a classic example of DOS using a biological molecule as a scaffold for potential drug design. Starting from the well-known benzopyran motif, the authors synthesize a collection of 22 compounds with a wide range of anti-tumor activities.
- Keaney GF, Johannes CW: Synthesis of an octahydroindolinone scaffold for a diversity-based chemical compound library. Tet Lett 2007, 48: 5411-5413.
- Blay G, Fernandez I, Molina E, Munoz MC, Pedroa JR, Vila C: Diastereoselective Michael addition of (S)-mandelic acid enolate to 2-arylidene-1,3-diketones: enantioselective diversity-oriented synthesis of densely substituted pyrazoles. Tet 2006, 62: 8069-8076.
-
** Garcia AB, Leßmann T, Umarye JD, Mamane V, Sommer S, Waldmann H: Stereocomplementary synthesis of a natural product-derived compound collection on a solid phase. Chem Comm 2006, 3868-3870.
This paper is notable because it successfully combines three desirable chemical features: an allylation reaction (a common way of introducing dense functionality), stereospecificity (essential for biologically active compounds), and solid-phase synthesis (greatly simplifying isolation and purification).
- Kashyap S, Hotha S: Stereoselective synthesis of a-glucosides from 3-O-propargyl protected glucal exploiting the alkynophilicity of AuCl3. Tet Lett 2006, 47: 2021-2023.
- Dai WM, Shi J: Diversity-oriented synthesis and solid-phase organic synthesis under controlled microwave heating. Comb Chem & High Thru Screen 2007, 20: 837-856.
-
* Noren-Muller A, Reis-Correa I Jr, Prinz H, Rosenbaum C, Saxena K, Schwalbe HJ, Vestweber D, Cagna G, Schunk S, Schwarz O, Schiewe H, Waldmann H: Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. PNAS 2006, 103(28): 10606-10611.
This paper clearly defines the philosophy behind BOS, and illustrates its potential power through the discovery of four novel classes of phosphatase inhibitors, two of which were the first for their respective phosphatases.
- Sohma Y, Taniguchi A, Yoshiya T, Chiyomori Y, Fukao F, Nakamura S, Skwarczynski M, Okada T, Ikeda K, Hayashi Y, Kimura T, Hirota S, Matsuzakid K, Kisoa Y: 'Click peptide': a novel 'O-acyl isopeptide method' for peptide synthesis and chemical biology-oriented synthesis of amyloid τ peptide analogues. J Pept Sci 2006, 12: 823-828.
- Umarye JD, Leßmann T, Garcia AB, Mamane V, Sommer S, Waldmann H: Biology-oriented synthesis of stereochemically diverse natural-product-derived compound collections by iterative allylations on a solid support. Chem Eur J 2007, 13: 3305-3319.
- Wyatt EE, Fergus S, Galloway WRJD, Bender A, Fox DJ, Plowright AT, Jessiman AS, Welchd M, Spring DR: Skeletal diversity construction via a branching synthetic strategy. Chem Comm 2006: 3296-3298.
- Thomas GL, Spandl RJ, Glansdorp FG, Welch M, Bender A, Cockfield J, Lindsay JA, Bryant C, Brown DFJ, Loiseleur O, Rudyk H, Ladlow M, Spring DR: Anti-MRSA agent discovery using diversity-oriented synthesis. Angew Chem Int Ed 2008, 47: 2808-2812.
-
** Wyatt EE, Galloway WRJD, Thomas GL, Welch M, Loiseleur O, Plowright AT, Spring DR: Identification of an anti-MRSA dihydrofolate reductase inhibitor from a diversity-oriented synthesis. Chem Comm 2008: 4962-4964.
This paper is the culmination of a drug development project from the Spring laboratory that used DOS to produce a new class of antibiotics and identify two members of that class with anit-MRSA activity. It is notable because it demonstrates the value of DOS across all phases of drug discovery, and may lead to a clinical validation of DOS.