Working with fellow MIT PhD candidate Tiffany Chen, I co-authored this NIH-style research proposal for the final project in my Fall 2008 Analysis of Biological Networks class at MIT. Download a PDF copy here.
Summary
Written by Brandon Russell
Platinum-based chemotherapies are among the most widely prescribed anti- cancer agents in the world, but the precise mechanism(s) by which they kill tumor-cells is widely unknown. A detailed understanding of this mechanism would provide new ways to reduce the side effects of existing chemotherapies, as well as provide new targets for chemotherapy development. Our proposal will employ a novel strategy to identify and quantify the exact cellular processes affected by these drugs, which will in turn allow us to construct a complete molecular mechanism of their action.
Abstract
Written by Brandon Russell
The platinum family of chemotherapeutics (including cisplatin, carboplatin, and oxaliplatin) are among the most widely prescribed drugs in modern oncology. They are especially common in the treatment of prostate cancer, which is currently the most diagnosed cancer among American males. Despite the ubiquity and extensive study into their chemistry and biology, the exact mechanism by which these drugs are cytotoxic to cancerous cells remains largely unknown. While much is known about their apoptotic effects on healthy cells, this is insufficient to explain their broadly effective anti-tumor activity, given the high frequency of apoptosis-deficient mutants found in most cancers. What studies have been conducted regarding the mechanisms of toxicity in cancerous cells have primarily relied on transcriptional profiling (microarray) technology, which is poorly suited to toxicity studies because of the wide array of posttranscriptional regulation that genes and proteins are known to be subject to. In order to guide rational design of new chemotherapeutics that augment both the positive anti-tumor and the negative side effects of existing platinum drugs, large volumes of translationally-based proteomic data are necessary. In this proposal, we utilize a novel procedure that is capable of selectively labelling and isolating newly synthesized proteins after a given time. Such temporal control allows for untargeted identification of only those proteins whose synthesis is augmented in response to platinum-family drugs, and the ease of the technique allows for its simultaneous application to a variety of treatment groups. By coupling this isolation technique with high precision mass spectrometry, we will generate a unique translational heat map showing the relative changes in protein levels in both healthy and cancerous cells exposed to cisplatin, carboplatin, and oxaliplatin. Subsequent analysis of the Worldwide Protein Data Bank will identify genes associated with these proteins, and will allow for the application of high quality sorting and enrichment algorithms such as Gene Ontology and Gene Set Enrichment. These methods will allow us to construct interactome maps that show the relation of the identified genes to known cellular processes. By dividing these maps into subnetworks using the Ingenuity software and assigning each subnetwork a score corresponding to the extent to which it was perturbed each drug, we will elucidate the molecular mechanism of cytotoxicity for each drug in both healthy and cancerous cells. This work will improve human health by providing novel strategies for increasing the efficiency and decreasing the side effects of both existing and subsequently developed chemotherapeutics.
Specific Aims
Written by Brandon Russell
The platinum family of chemotherapeutics, which includes the parent compound cisplatin and its derivatives carboplatin and oxaliplatin, are among the most commonly prescribed anticancer agents, with global sales of over $670 million in 2004 [1]. Platinum family compounds are especially common in the treatment of prostate cancer, one of the most common cancers in American males. Despite their widespread use, the molecular mechanisms of their anti-tumor activity remains largely unknown. What work has been done has primarily relied on traditional transcriptional profiling approaches (microarrays), which are poorly suited to toxicological and pharmaceutical studies because they do not capture the spectrum of proteomic changes that underlie phenotypic cellular responses. A detailed understanding of the mechanism of action of platinum chemotherapeutics would provide insight into the development of resistance and help guide the rational design and synthesis of improved derivatives. Toward the goals of expanding our knowledge of existing chemotherapeutics and improving our ability to develop new ones, we propose a novel, untargeted, translational profiling method to elucidate the mechanisms of anti-prostate cancer activity by cisplatin, carboplatin, and oxaliplatin.
Specific Aim 1: Create translational profiles of the effect of platinum chemotherapeutics on human prostate cells.
As the building blocks of nearly all biological stuctures, proteins are the basic functional unit of cellular processes; changes in a cell's appearance, behavior, or fate can always be traced back to changes in proteins. Additionally, the role of posttranslational modifications in modulating cellular phenotypes has become increasingly studied in recent years. Together these points underscore the need for detailed proteomic data to understand cellular mechanisms. Because death by any mechanism represents a change in response to a perturbation, proteins that are synthesized after drug delivery and whose synthesis is modulated (upregulated or downregulated) by chemotherapy are of primary interest. Given the analogue nature of protein levels and modifications, any given stimulus will produce a unique change in the protein "landscape" of the cell. Our first specific aim is to use a recently developed labelling and isolation protocol (described in detail later) that is specific for newly translated proteins combined with high-sensitivity mass spectrometry to produce a translational map of the response to cisplatin, carboplatin, and oxaliplatin in human prostate tissue. By subjecting primary samples from both healthy and cancerous prostates to identical treatment and comparing untreated samples under otherwise identical conditions, we will generate extensive proteomic data with full inclusion of both variables (treated vs. untreated and healthy vs. cancerous).
Specific Aim 2: Identify genes coding for the identified proteins and create interactome maps that define each drug's molecular mechanism of action.
High volume proteomic data is essential for a complete mechanistic description of cellular phenotype, and requires significant processing to identify and isolate pertinent trends. Publicly searchable protein databases (such as the Worldwide Protein Data Bank) will be used to identify the genes coding for the proteins identified. Heat maps will then be constructed for each chemotherapeutic, showing the relative change in expression of each protein in healthy and cancerous prostate cells. To better highlight mechanistic information, gene ontology enrichment analysis and Gene Set Enrichment Analysis will be performed according to established protocols, thereby condensing the results into meaningful phenotypic categories. Using this information, an interactome will then be constructed by querying the Ingenuity database according to established protocols. Subsequent division of this interactome into subnetworks will identify cellular processes most strongly modulated by platinum compounds, which will provide the basis for a complete mechanism of action and will guide the search for improved chemotherapeutics that both enhance the potency and ameliorate the side effects of existing drugs.
Background & Significance
Written by Tiffany Chen
Prostate Cancer
Prostate cancer is the second leading cause of male deaths from cancer in the United States, right behind lung cancer. Typically, it impacts older men; African American men older than 60 are especially at high risk, and it is rarely found in men younger than 40 [2]. It has the highest rates of incidence in the United States and then Europe, and is the least common in South and East Asia [3]. There is speculation that this increase in incidence rate could be because of better detection technologies, and perhaps even some overdiagnosis.
Prostate cancer only affects men, since the prostate is a gland of the male reproductive system. Classified as an adenocarcinoma, the cancer occurs when the normal gland cells mutate after continual damage and avoid apoptosis. Typically the cancer cells linger in the peripheral zone of the prostate gland, but eventually they begin to multiply and spread, leading to tumor growth. The direct causes of prostate cancer are unknown though relationships have been established through high fat intake and increased testosterone levels [2].
Methods of screening include digital rectal exams, urinalysis, biopsy, and prostate specific antigen (PSA) blood tests, which can detect very minute amounts of antigen. This high sensitivity occasionally leads to detection of small non-life threatening cancers, hence overdiagnosis. Since prostate cancer grows slowly and usually impacts older males, some of these men, positively diagnosed with PSA, tend to pass away from other causes instead of prostate cancer [2]. Treatment options range from surgery, radiation therapy, chemotherapy, and hormonal therapy. Our interests lie within the use of platinum based chemotherapeutics to treat prostate cancer [4]. The actual mechanism of how this class of chemotherapeutics specifically kills cancer cells is unknown. Further exploration of this can lead to a full or perhaps better understanding of its mechanism of action.
Platinum Chemotherapeutics
Cisplatin can almost be referred to as the penicillin of chemotherapies. Barnett Rosenberg first discovered its use in 1965 when he was studying the effects of electric fields on Escherchia coli growth [5]. His experiments were set up using platinum electrodes and aqueous NH4Cl as an electrolyte. He discovered that under the electric field, bacterial cell division stopped and the bacteria formed long filaments that were 300 times the normal length. After performing further studies he determined that platinum complexes coming off of the platinum electrodes as electrolysis products were responsible for preventing cell division. In the bacterial studies, it was found that two types of complexes were formed, charged and neutral complexes. The charged complexes were bacteriocidal, whereas the neutral complexes only prevented cell division. Neutral complexes were formed from charged complexes through exposure to ultra violet light, in both cis- and trans- formations. Depending on the valence state of the platinum (+2 or +4), complexes would either be square planar or octahedral forms. From these studies, the most effective compound in causing filament formation was cis- diamminedichloroplatinum(II) (cisplatin), also known as "Peyrone's Salt" first synthesized in 1845 by Michel Peyrone [6]. Trans complexes did not perform as well in increasing filament formation.
These complexes were linked to the activation of lysogenic bacteria, causing bacterial lysis. From this, it was determined that the platinum complex was having an interaction with DNA. It was also noted that at very low concentrations of cisplatin, prevention of bacteria cell division was observed without marked toxicity [7]. This led to heightened interest in using cisplatin as an anti-cancer drug. These neutral complexes were shown to be useful as anti-cancer drugs in studies on sarcoma 180 and leukemia in mice [8]. Clinical trials were performed soon after, and it has been shown that kidney toxicity is the main limitation on cisplatin dosage.
Cisplatin is also known to be synergistic with other anti-cancer drugs and widely used in combination therapy [7]. In the late 1970s, cisplatin was finally approved for clinical use as an anti-tumor drug and has been shown to be effective in treating testicular, ovarian, head, and neck cancers among others. Certain tumors are also known to be intrinsically resistant to cisplatin, such as non-small cell cancer [1]. Concerns over nephortoxicity and ototoxicity lead to the synthesis of other platinum derivatives including carboplatin and oxaliplatin.
Molecular Mechanism of Action
The basic molecular mechanism of action of cisplatin binding to DNA is known, but understanding why cisplatin causes higher cell death in cancerous cells as opposed to normal cells still remains largely unknown. Cisplatin reacts more readily with DNA in its hydrolyzed form when a water molecule displaces a chloride ion [1]. Because if its structure, the chloride ions on transplatin are replaced quicker with water molecules and leading to faster reactions with other molecules rendering it inactive. Therefore, a high enough concentration of transplatin is never able to actually make into a cell to react with the DNA [9]. Cisplatin is mainly inactive in the blood stream because there is an increased concentration of chloride ions in the blood, thus preventing replacement of chloride ions. In this form, cisplatin is still vulnerable to reaction with other proteins in the blood, especially ones containing cysteines and/or thiols, such as human serum albumin. Once the protein is bound to cisplatin, the drug is deactivated, and therefore this could be one of the reasons behind cisplatin resistance.
Cisplatin enters cells through diffusion past the cell membrane, and once inside the cell becomes hydrolyzed because intracellular chloride concentration is lower than in the blood. Once hydrolyzed, cisplatin can no longer diffuse out of the cell. Cisplatin is charged and reactive to nucleophilic groups of oxygen, nitrogen and sulfur; these have unpaired electrons, which can bind with platinum substituting for the chloride ion(s). Therefore, cisplatin is reactive to not only DNA but also other molecules such as glutathione, which allows it to be transported out of the cell. This mechanism could also be another component to the increase in cisplatin resistance [10]. Once cisplatin is able to make it into the nucleus in its active form, it tends to react with GC-rich regions of DNA [7].
Cisplatin can react at two sites on a given base, especially with purines (the N7 atoms of guanine and adenine) or two sites of two different bases. This allows for crosslinking of two strands of the double helix DNA, making it similar to alkylating agents, or it can crosslink two neighboring bases on a single strand. It is known that cisplatin does not react with the sugar-phosphate backbone of DNA, only the bases, and it also does not intercalate between the bases. 1,2-dGpG and 1,2-dApG intrastrand crosslinks make up 90% of DNA adducts [7]. This crosslinking forms covalent DNA adducts causing the DNA to bend and widen at the minor groove. This structure has been confirmed through x-ray crystallography and NMR spectroscopy, which showed that the platinum ion lies in the minor groove of the DNA [11]. It is proposed that this change in structure serves as a recognition point for certain proteins that bind and process damaged DNA [12].
Under this genotoxic stress, there are various proteins that are up regulated such as DNA repair proteins, p53 regulatory pathway proteins, and transcription factors. Depending on how these proteins process the cisplatin caused DNA adduct the cell will either be repaired or under go apoptosis. Cisplatin adducts are shown to inhibit both transcription, by blocking RNA polymerase II, and also DNA synthesis. It has been shown in vivo that cisplatin caused DNA adducts are repaired with poor efficiency, thus leading its use as an anti-cancer drug [13]. In human cells, the main repair pathway used to fix cisplatin adducts in DNA is the nucleotide excision repair (NER) pathway, typically used to remove pyrimidine dimers caused by Ultraviolet light.
The reason why cisplatin adducts are poorly repaired lies in the class of cellular proteins that bind to sites that are structurally similar to the DNA adduct formed by cisplatin. These proteins known as high-mobility-group (HMG) proteins end up binding to the cisplatin adducts and block repair proteins from recognizing the lesion. Without repairing the DNA, the cell eventually undergoes apoptosis. Under endogenous levels of HMG, it appears that cells are able to repair the DNA adduct, but under excess concentration of HMG cell repair is inhibited [13]. The up regulation and down regulation of these proteins in cancerous versus normal cells will be one of our interests in this project.
Cisplatin adducts have been linked to triggering apoptosis through many different mechanisms. These include G2 cell-cycle arrest, induction of p53 and p73 regulatory proteins, expression of Bcl-2 family proteins, and mismatch repair, among many others [13,14]. Other studies have linked cisplatin DNA adduct caused cell death to necrosis [15,16]. Alternative platinum based chemotherapeutics have also been somewhat studied. Carboplatin interacts with DNA in basically the same approach as cisplatin, except that it has different kinetics [17]. Oxaliplatin on the other had is different it its function from cisplatin and carboplatin, but it is not very well characterized [18]. As evident, the mechanism of cell death caused by cisplatin is not very well known, but our novel approach by measuring varying protein concentrations caused by platinum based chemotherapy exposure at the systematic level will help us identify different networks key to platinum-based cell destruction.
Bioorthogonal Noncanonical Amino Acid Tagging
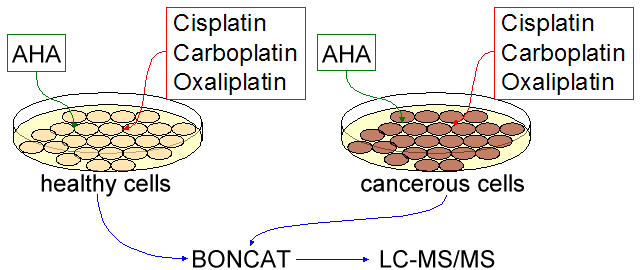
A strategic technology we are utilizing in our approach is bioorthogonal noncanonical amino acid tagging (BONCAT). This method uses the noncanonical amino acid, azidohomoalanine (AHA), which is incorporated into proteins in place of methionine when the cells are grown in a media lacking methionine. Therefore, BONCAT technology is able to identify about 94% of the mammalian proteome, which include proteins that have at least one methionine residue given that it is not an N-terminal methionine subjected to post-translational removal [19]. This tool has already been shown to selectively identify newly synthesized proteins in mammalian cells [20].
Because we want to approach this problem at a systematic level, using BONCAT will allow us to view the entire proteome of prostate cells (normal and cancerous) under platinum based chemotherapeutic stress. Knowing the behavior of protein expression due to this stress will allow us to trace back and see which genes are being turned on, but at the view point from protein levels and not mRNA levels. Protein levels are a better representation of the activation of certain pathways in the cells than mRNA, since translation can be up regulated or down regulated depending on modifications after transcription. This in turn can elucidate which networks or pathways are playing major roles in cell death due to platinum adduct formation.
Research Design & Methods
Specific Aim 1: Create translational profiles of the effect of platinum chemotherapeutics on human prostate cells.
Written by Brandon Russell
Rationale: Despite their widespread use in a variety of human cancers, platinum family chemotherapeutics have poorly defined modes of action at the molecular level. A thorough understanding of the cytotoxic mechanism of such drugs (against both healthy and cancerous tissues) would aid the search for more effective chemotherapies with fewer side effects. Transcriptionally based approaches are poorly suited to such studies as they neglect the spectrum of post- transcriptional (such as modulated translation rate through modified tRNAs) and translational (such as modulated stability through phosphorylation) modifications that are essential for cellular responses. By employing a newly developed technique that can selectively label and isolate proteins synthesized after a given time point, we can capture the full spectrum of such changes induced by platinum chemotherapeutics in both healthy and cancerous cells. This work will yield a wealth of highly accurate, unique proteomic data.
Methodology: The analytical method underlying our proposal is known as bioorthogonal noncanonical amino acid tagging, and was described and optimized by Dietrich et al. in 2007 [19,20]. The method utilizes the unnatural amino acid azidohomoalanine (AHA), which is a methionine surrogate bearing an azide functional group as part of the sidechain. The azide group is unique in biology because it is almost completely abiotic, occurring only in a few species of algae but displaying no toxicity in E. coli, yeast, or mammals [24]. Kiick et al. demonstrated that E. coli grown in methionine-free medium supplemented with AHA incorporated AHA without bias, without perturbation in translational rate or fidelity, and without cytotoxicty. The azide group is well known in "click chemistry" because of the Huisgen cyclization, which combines an azide with a terminal alkyne in the presence of a copper(I) catalyst to produce a stable 1,2,3-triazole species. The reaction proceeds to >90% completion on the time scale of hours at 4°C in aqueous solvent with no toxic side products, and as such is ideally suited for use in biological systems [25]. The favorability of the reaction allows for extensive functionalization of the terminal alkyne, including the addition of fluorophores, Flag tags, and affinity handles. The result is a method capable of selectively labelling and separating newly synthesized proteins, which can then be subjected to traditional mass spectrometry identification. The full analytical scheme is illustrated in Figure 1 - schematic flowchart of bioorthogonal noncanonical amino acid tagging analysis [19].
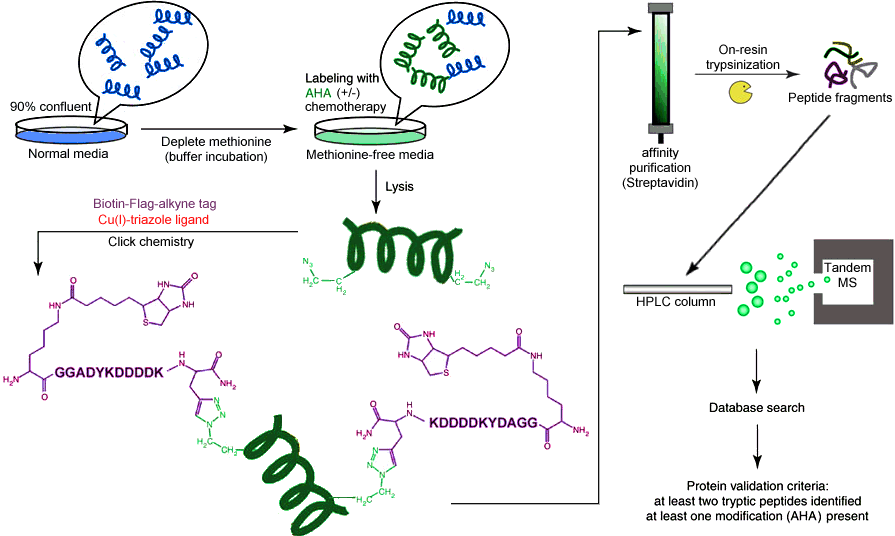{kind=link}
Materials: Primary healthy human prostate cells and appropriate medium will be obtained from Lonza. Primary human prostate cancer cells will be obtained from collaborators, with collection and record keeping protocols established such that we cannot subsequently access direct or indirect identifiers, thereby qualifying this project for HHS Protection of Human Subjects Regulations Exemption 4. Custom methionine- free medium will be obtained from Millipore. AHA will be custom ordered from Arcos or synthesized in-house using commercially available reagents (Aldrich) and an established scheme [22,23], whichever is deemed more efficient. Reagent grade copper(I) bromide will be obtained from Aldrich, and the triazole ligand will be synthesized in-house using commercially available reagents (Aldrich) and an established scheme [26]. A custom alkyne ligand bearing a biotin affinity handle and a trypsin-cleavable Flag epitope will be obtained from GenScript Corporation. Cisplatin, carboplatin, and oxaliplatin will be obtained from Aldrich. All other standard bioanalytical reagents will be obtained from appropriate commercial sources. Peptide analysis will be performed via HPLC coupled to an electrospray-ionization ion-trap tandem mass spectrometer.
Experimental Design: Cells will be grown according to established protocols in standard medium. When cells are 90% confluent, they will simultaneously be transferred to buffer solution and incubated for 30 minutes to deplete the free amino acid concentration. They will then be replated at optimum density on methionine-free medium supplemented with AHA, and each category of cells (healthy and cancerous) will be divided into four treatment groups: untreated (control), cisplatin, carboplatin, and oxaliplatin. Chemotherapeutics will be added at the same time as AHA, and concentrations will be scaled to match the standard human dose for each drug. Treated cells will be grown until no further cytotoxicity is observed, and untreated cells will be grown until replating is necessary or for a time matching the shortest growth of a treated colony, whichever is less. After incubation, cells will be collected and lysed according to the established protocol [19], and the soluble fraction will be isolated. Isolated fractions will be mixed with the alkyne ligand, Cu(I) bromide, and triazole ligand, and the reaction will be allowed to proceed to completion (18h at 4°C). Following the ligation, excess ligand will be removed via gel filtration and the ligation will be confirmed by dot blot detection/quantification of biotinylated (newly synthesized) proteins. Following confirmation, proteins will be isolated via affinity chromatography on a NeutrAvidin column (16h at 4°C), then trypsinized on-resin (overnight at 37°C). Trypsinized peptides will be isolated from the resin by microcentriguation in a spin column. Samples will be collected at appropriate points to confirm key steps by dot blot analysis. The resulting peptide fractions will be separated by reversed-phase HPLC and identified by tandem mass spectrometry using the Mascot search engine augmented to account for possible side products from incomplete tagging and/or cleavage. The resulting relative protein levels will be normalized by soluble fraction volume, allowing for untargeted identification of significantly affected proteins [26]. All procedures will be performed in triplicate, and the overall scheme is illustrated in Figure 2 - visual summary of the experimental design for Specific Aim 1.
{kind=link}
Potential Pitfalls: Some laboratories have reported low yields for the click chemistry ligation, owing primarily to the insolubility of the Cu(I) bromide in water (Erik Wilker, personal communication). If this proves to be the case in our preliminary results, there is an alternative ligation procedure that has been shown to have equivalent sensitivity and kinetics [27]. This alternative procedure eliminates the need for a catalyst and incorporates the alkyne ligand into a ring-strained difluorocyclooctyne (DIFO). If the copper catalyzed protocol above is insufficient, we will synthesize in-house a custom DIFO ligand using commercially available reagents (Aldrich) and an established scheme [27]. This ligand will possess the same functionalities as described above, and the rest of the protocol will remain unchanged. Several laboratories have also reported unacceptably low yields from the affinity purification and subsequent on-resin trypsinization (Michael Yaffe, personal communication). While we are confident in our ability to optimize the step via changes to the lysis and cleavage protocols, we are prepared to implement an alternative data acquisition strategy. In the event that the above strategy proves unfeasible, we will obtain a custom alkyne ligand bearing a fluorophore from a commercial source (Invitrogen). The ligation reaction will be carried out as described above using the new ligand, and the cells will be lysed according to standard protocol. The soluble fraction will then be subjected to two-dimensional gel electrophoresis, and the fluorescent spots will be excised and analyzed by the same LC- MS/MS protocol as described above. This method is less sensitive and significantly more time- and labor-intensive than the above, but is a well established technique that is essentially guaranteed to succeed. It is important to note that these two pitfalls and their solutions are not mutually exclusive. Therefore, if both the ligation step and the subsequent affinity purification step prove to be inefficient, we will synthesize in-house a custom DIFO ligand bearing a fluorophore and apply the 2D gel based approach described above.
Summary: The molecular mechanism of cytotoxicity by platinum drugs in cancerous cells is poorly defined. Effective rational design and development of improved drugs that increase anti-tumor activity (particularly in cisplatin-resistant cancers) and/or decrease collateral toxicity requires a detailed understanding of this mechanism. Transcription- based approaches have been largely ineffective at this task because they do not account for a vast array of post-transcriptional regulatory steps that separate mRNA from functional proteins. We propose a novel, untargeted, translation-based approach to identify proteins whose levels are significantly modulated in prostate cells exposed to clinical levels of cisplatin, carboplatin, and oxaliplatin. This project will generate a large volume of unique proteomic data for each drug, improving both the basic science understanding of chemotherapy and the clinical search for improved drug targets.
Specific Aim 2: Identify genes coding for the identified proteins and create interactome maps that define each drug's molecular mechanism of action.
Written by Tiffany Chen
Rationale: In order to obtain a systematic view of the effects of cisplatin, carboplatin, and oxaliplatin on prostate cancer cells and normal cells, changes in protein expression levels must be first identified and then fully mapped out. Comparisons will be drawn between cancerous cell and normal cell protein levels and also with treated and untreated cells. By analyzing these differences, networks that play major roles in response to these chemotherapeutics can be identified. To perform these analyses, full use of bioinformatics will be employed to identify proteins, their respective genes, and interactions with other proteins or genes.
Identification of LC-MS/MS products: Eight sets of protein data would be obtained through the LC-MS/MS. These sets include treated (with cisplatin, carboplatin, and oxaliplatin) cancer cells and healthy cells and untreated cells, with the untreated cells being our controls. Protein amounts from the treated cells will be compared to the untreated cells to obtain relative concentration levels. The products from our LC- MS/MS scan will be determined using Mascot.
Primary organization of heat maps: A preliminary set of heat maps will be compiled using the values of the relative protein levels from LC-MS/MS. Since the names of the proteins are known, these identified proteins and their respective values can be made into a matrix and graphically displayed as a heat map. From this heat map, up regulated and down regulated proteins will be selected for further analysis. Determine specific proteins and their respective genes: Once specific proteins are identified, their functional roles can be determined through publicly searchable protein databases, such as the Worldwide Protein Data Bank, Uniprot, which consists of Swiss- prot and TrEMBL, and GenBank among many others. These databases will also contain information about the specific genes coding for the proteins. Through this database search we can trace back the gene coding for the protein, the other proteins that interact with the protein, and role in certain pathways.
Secondary organization of heat maps: In this set, two heat maps of selected protein levels for each drug, one for treatment of cancer cells and the other for treatment of healthy cells, are created in comparison to the uncreated cancer cells and healthy cells, respectively. A total of 6 heat maps will be organized to show the relative differences in the selected protein levels, including both protein name and its respective gene. From there, specific genes can be selected for further analysis with gene ontology. Gene Ontology enrichment analysis and network maps: By studying the ontology of the genes that code for differentially expressed proteins, specific gene sets can be determined. These gene sets can then undergo gene ontology enrichment analysis, using GO Miner, to categorize the genes that change in protein expression due to certain platinum-based chemotherapy exposure. Furthermore, Gene Set Enrichment Analysis (GSEA) will be used to determine the expression signatures of groups of genes that perform similar functions. These two programs serve as filters to determine specific genes of interest. Afterwards, the Ingenuity knowledge base will determine the connections between the genes, and hence suggest possible connections between proteins. Using these connections, network maps will be created to illustrate significant protein interaction. From these, sub-networks based on specific pathways can also be outlined.
Potential Pitfalls: Since this project aims to identify various protein levels at the systematic level, without choosing explicit proteins ahead of time to study, finding patterns within protein expression levels may be difficult. If no specific patterns can be identified after gene ontology enrichment analysis is performed, the focus can be shifted to proteins that have been known to modulate according to platinum-based chemotherapy treatment.
Summary: Efficient and logical organization of the large volume of data generated in Specific Aim 1 is essential if we are to extract meaningful conclusions. By comparing the relative levels of each identified protein in treated and untreated healthy and cancerous cells, a unique heat map for each drug's effect on both healthy and cancerous tissue will be generated. Using the Worldwide Protein Data Bank to equate identified proteins with known genes, we open up our data to a wide variety of powerful analytical tools, including Gene Ontology and Gene Set Enrichment. By applying these sorting and clustering tools, we will identify broad categories of cellular functions that are perturbed by each chemotherapy, including those categories that are differentially affected in healthy and cancerous cells. By using the Ingenuity software, we will assemble these categories into defined subnetworks, each with a varying level of susceptibility to platinum chemotherapeutics. This will allow us to construct a complete molecular mechanism of platinum drugs in both healthy and cancerous cells, which will in turn improve human health by guiding future design of improved chemotherapeutics with great anti-tumor activity and/or ameliorated side effects.
Literature Cited
- Alderden, R.A., M.D. Hall, and T.W. Hambley, The Discovery and Development of Cisplatin. Journal of Chemical Education, 2006. 83(5): p. 728-734.
- Nanda, R., Prostate Cancer, in Medline Plus. 2006, NIH.
- IARC Worldwide Cancer Incidence Statistics-Prostate. 2001 [cited 2008-11-23].
- Zhang, W. and M. K. C. Balaji, Role of Apoptosis Inducing Factor in Prostate Cancer Cell Apoptosis Induced by Cisplatin, in New England Section of the American Urological Association. 2006: Providence, Rhode Island.
- Rosenberg, B., L.V. Camp, and T. Krigas, Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature, 1965. 205: p. 698-699.
- Peyrone, M., Ann Chemie Pharm, 1845. 51: p. 129.
- Lippert, B., Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. 1999: Helvetica Chimica Acta.
- Rosenberg, B., et al., Platinum Compounds: a New Class of Potent Antitumor Agents. Nature, 1969. 222: p. 385-386.
- Sadler, P.J. and Z. Guo, Metal Complexes in medicine: Design and mechanism of action. Pure & Appl. Chem., 1998. 70(4): p. 863-871.
- Stordal, B. and M. Davey, Understanding Cisplatin Resistance Using Cellular Models. IUBMB Life, 2007. 59(11): p. 696-699.
- Reedijk, J., The mechanism of action of platinum anti-tumor drugs. Pure & Appl. Chem., 1987. 59(2): p. 181-192.
- Wozniak, K. and J. Blasiak, Recognition and repair of DNA-cisplatin adducts. Acta Biochimica Polonica, 2002. 49(3): p. 583-596.
- Jordan, P. and M. Carmo-Fonseca, Molecular mechanisms invovled in cisplatin cytotoxicity. Cellular and Molecular Life Sciences, 2000. 57: p. 1229-1235.
- Siddik, Z.H., Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene, 2003. 22: p. 7265-7279.
- Lieberthal, W., V. Triaca, and J. Levine, Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. encrosis. AJP - Renal Physiology, 1996. 270(4): p. 700-708.
- Fuertes, M.A., et al., Cisplatin Biochemical Mechanism of Action: From Cytotoxicity to Induction of Cell Death Through Interconnections Between Apoptotic and Necrotic Pathways. Current Medicinal Chemistry, 2003. 10(3): p. 257-266.
- Knox, R.J., et al., Mechanism of Cytotoxicity of Anticancer Platinum Drugs: Evidence that cis-Diamminedichloroplatinum(II) and cis-Diammine-(1,1- cyclobutanedicarboxylato)platinum(II) Differ Only in the Kinetics of Their Interaction with DNA. Cancer Research, 1986. 46: p. 1972-1979.
- Raymond, E., et al., Cellular and Molecular Pharmacology of Oxaliplatin. Molecular Cancer Therapeutics, 2002. 1: p. 227-235.
- Dieterich, D., et al., Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nature Protocols, 2007. 2(3): p. 532-540.
- Dieterich, D., et al., Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). PNAS 2006. 103(25): p. 9482-9487.
- Link, A.J. et al. Preparation of the functionalizable methionine surrogate azidohomoalanine via copper-catalyzed diazo transfer. Nature Protocols 2007, 2: 1879- 1883.
- Link, A.J. et al. Synthesis of the functionalizable methionine surrogate azidohomoalanine using Boc-homoserine as precursor. Nature Protocols 2007, 2: 1884- 1887.
- Griffin, R.J. The medicinal chemistry of the azido group. Progress in Medicinal Chemistry 1994, 31: 121-232.
- Link, A.J. et al. Presentation and detection of azide functionality in bacterial cell surface proteins. Journal of the American Chemical Society 2004, 126: 10598-10602.
- Wang, Q. et al. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. Journal of the American Chemical Society 2003, 125: 3192-3193.
- Saghatelian, A. et al. Assignment of Endogenous Substrates to Enzymes by Global Metabolite Profiling. Biochemistry 2004, 43: 14332-14339.
- Baskin, J.M. et al. Copper-free click chemistry for dynamic in vivo imaging. PNAS 2007, 104: 16793-16797.